Download præsentationen
Præsentation er lastning. Vent venligst

1
”Genetisk rådgivning af kræftpatienter og deres pårørende nu og i fremtiden”
Mette Wilhjelm og Susanne Hein Onkologisk Klinik/genetisk afdeling Rigshospitalet FSK landskursus 2014 Tidl har pernille hertel undervist i genetik, pernille er min daglige samarbejdspartner. Noget vil være en gentagelse men jeg vil forsøge at have mere fokus på sygeplejen eller assistent opgaverne

2
HVEM ER VI Mette Wilhjelm: Onkologisk sygeplejerske, Rigshospitalet
Arbejdet de sidste 8 år på Mammae Teamet, Afdeling 4262 Speciale i brystkræft Specialuddannelsen i kræftsygepleje Arbejdet med genetisk rådgivning af brystkræftpatienter i 2 år Susanne Hein: Sygeplejerske, genetisk vejleder, Klinisk Genetisk Klinik, Rigshospitalet Derudover rådgiver på Kræftlinjen Jeg hedder mette wilhjelm

3
INDHOLD I DAG Formål ved genetisk rådgivning
Genetisk rådgivning - hvor? Henvisning til genetisk rådgivning Kræft, en arvelig sygdom Arvelige genfejl Arvelig bryst og æggestokkræft Kriterier for henvisning Lynch syndrom HNPCC Andre arvelige kræftformer Cases

4
FORMÅL MED GENETISK RÅDGIVNING
At sikre alle familier med arvelig disposition til cancer et tilbud om: Genetisk udredning, Risikovurdering, Rådgivning Molekylærgenetisk diagnostik

5
FORMÅL MED GENETISK RÅDGIVNING
At finde de patienter som har en stor risiko for at få kræft. At målrette behandling til patienter der har en genetisk fejl At forlænge levetiden hos patienter med arvelig kræft Tidlig opsporing af ny kræftsygdom Forebyggelse af ny kræftsygdom Undgå at de i fremtiden ikke kommer med en kræft, som ikke kan helbredes Give patientens nære slægtninge mulighed for: Genetisk rådgivning Finde de pårørende, som skal i et specielt kontrolprogram, hvis de er i højere risiko end normalbefolkningen for at få kræft.

6
GENETISK RÅDGIVNING – HVOR?
Henvisninger fremsendes til den regionale klinisk genetiske afdeling: Klinisk Genetisk Klinik, Rigshospitalet Onkologisk Klinik mammae teamet, 4262, Rigshospitalet Kennedy Centret, Glostrup, Rigshospitalet Klinisk Genetisk Afdeling, Odense Universitetshospital Klinisk Genetisk Afdeling, Vejle Sygehus Klinisk Genetisk Afdeling, Aarhus Universitetshospital, Skejby Klinisk Genetisk Afdeling, Aalborg Sygehus Man kan få genetisk rådgivning i Danmark, som der fremgår ad slides

7
HVORDAN BLIVER MAN HENVIST TIL GENETISK RÅDGIVNING?
Via sin praktiserende læge (evt. på opfordring af andet familiemedlem) Via speciallæge Via sygehuset Via andre genetiske afdelinger i landet Henvisning at personer/familier mistænkt for arvelig disposition til cancer mammae og/eller ovarii til genetisk udredning og rådgivning modtages fra almen praksis, hospitalsafdelinger og speciallægepraksis.
 Via speciallæge. Via sygehuset. Via andre genetiske afdelinger i landet. Henvisning at personer/familier mistænkt for arvelig disposition til cancer mammae og/eller ovarii til. genetisk udredning og rådgivning modtages fra almen praksis, hospitalsafdelinger og. speciallægepraksis.")
8
HVORDAN FÅR MAN MISTANKE OM AT KRÆFT KAN VÆRE EN ARVELIG SYGDOM?
Mange tilfælde af cancer (med samme diagnose) i familien Debut af cancersygdom før 40 – 45 års alderen Flere familiemedlemmer med flere forskellige cancersygdomme
 i familien. Debut af cancersygdom før 40 – 45 års alderen. Flere familiemedlemmer med flere forskellige cancersygdomme.")
9
TYPER AF ARVELIGE GENFEJL
Sygdomsfremkaldende Fejlen sidder et kritisk sted på genet Genet fungerer ikke Ukendte Vurdering af betydning Vurdering af forekomsten Funktionelle studier (tager meget lang tid) Ubetydende genfejl Fejlen sidder et sted på genet, hvor det ikke er kritisk, ikke nødvendigvis ændrer på afkodningen.
 Ubetydende genfejl. Fejlen sidder et sted på genet, hvor det ikke er kritisk, ikke nødvendigvis ændrer på afkodningen.")
10
HVAD KRÆVER DET AF PATIENTEN FØR GENETISK RÅDGIVNING
Stort arbejde at udfylde slægtsbog Måske nødvendigt at kontakte familiemedlemmer, som de sjældent ser, for at få de nødvendige oplysninger Får en viden om slægtninge (skæbner)som de ikke vidste på forhånd - blandt andet årsager til død Bliver bekendt med slægtninge som de ikke vidste fandtes i familien
som de ikke vidste på forhånd - blandt andet årsager til død. Bliver bekendt med slægtninge som de ikke vidste fandtes i familien.")
11
EPIDEMIOLGI INDEN FOR BRYSTKRÆFT
Cirka 4500 nye tilfælde af brystkræft i DK om året Nordcan 2011 Tidlig pubertet Høj alder ved første fødsel Sen menopause Hormontilskud efter overgangsalderen Alkohol Overvægt efter overgangsalderen Fysisk inaktivitet Natarbejde Ioniserende stråling Ethylenoxid Celleforandringer i brystet Genfejl Visse meget sjældne sygdomme/syndromer 5-10% Brystkræft

12
BRYSTKRÆFT OG GENFEJL Genfejl nedarves fra generation til generation
Opstår sjældent ved undfangelsen Sjældent nedarves mere end en betydende genfejl Fejl i et gen betyder ikke noget for den enkelte celle, men hvis den anden genkopi rammes af anden fejl, er reparationsmekanismen ikke fungerende Ved næste mutation kan cellen ikke repareres, hvilket kan føre til cancer Cancer skyldes en ophobning af mutationer i gener forårsaget af genetiske faktorer og/eller miljøfaktorer. En medfødt mutation i et enkelt gen kan alene føre til en meget høj risiko for cancer mammae og cancer ovarii, som det er tilfældet ved højpenetrante mutationer i f.eks. BRCA1 og BRCA2 . Risikoen for cancer mammae og cancer ovarii kan dog også være øget som følge af lavpenetrante mutationer. Hos patienter med cancer mammae har påvisning af en højpenetrant mutation i BRCA1/2 en prædiktiv værdi, idet disse patienter har en meget høj risiko for en ny primærtumor i mammae og ovarier. Kilde DBCG Hver familie har deres egen genfejl Brystkræft

13
BRCA1 BRCA2 17q21 13q12 BETYDENDE GENER Sekventeret i 1994
BRCA1 blev lokaliseret til kromosom 17's lange arm i 1990, og genet blev isoleret i 1994 (3,4). BRCA2 blev lokaliseret til kromosom 13 i 1994, og indenfor 1 år var genet isoleret (5). Kvinder, der er bærere af medfødte BRCA1- eller BRCA2-mutationer, har en meget høj livstidsrisiko for cancer mammae (også bilateralt) og/eller cancer i ovarier/salpinges/peritoneum. Alle de kvindelige bærere får dog ikke cancer, ligesom risikoen kun er let forøget hos mænd. Sekventeret i 1994 Sekventeret i 1996 22 exons koder for et protein med aminosyrer 27 exons koder for et protein med aminosyrer Cancer mammae et ovarii Cancer mammae et ovarii be 09.99
. BRCA2. blev lokaliseret til kromosom 13 i 1994, og indenfor 1 år var genet isoleret (5). Kvinder, der er bærere af. medfødte BRCA1- eller BRCA2-mutationer, har en meget høj livstidsrisiko for cancer mammae (også. bilateralt) og/eller cancer i ovarier/salpinges/peritoneum. Alle de kvindelige bærere får dog ikke cancer, ligesom risikoen kun er let forøget hos mænd. Sekventeret i Sekventeret i exons koder for et protein. med aminosyrer. 27 exons koder for et protein. med aminosyrer. Cancer mammae et ovarii. Cancer mammae et ovarii. be")
14
DOMINANT ARVELIG BRYSTKRÆFT
BRCA2 Andre Eks:CDH1,TP53, PTEN, RAD51C, Ukendt 50% BRCA1 25% BRCA1-mutationer er påvist hos 15 – 20% af patienter med en familiehistorie med cancer mammae, og 60 – 80% af patienter med både cancer mammae og cancer ovarii i familien (6, 7). Livstidsrisikoen for cancer mammae har hos bærere af BRCA1-mutationer varierer mellem 56% og 87% i forskellige undersøgelser (8–10). Den mediane alder på diagnosetidspunktet for cancer mammae er 42 år hos BRCA1-mutationsbærere, hvilket er 20 år tidligere end hos patienter med sporadiske tumorer (11). Livstidsrisikoen for cancer ovarii varierer mellem 20 og 60% (10,12). BRCA2-mutationer er formentlig ansvarlige for en mindre del af arvelig disposition til cancer mammae og cancer ovarii end BRCA1-mutationer. Den mediane alder på diagnosetidspunktet for cancer mammae er 49 år, hvilket er højere end hos bærere af BRCA1-mutationer men lavere end for kvinder med sporadisk cancer mammae (13). Livstidsrisikoen for cancer mammae varierer i forskellige undersøgelser mellem 28 og 85% (14). Livstidsrisikoen for cancer ovarii er øget hos bærere af BRCA2- mutationer (10 – 27%). Hos mandlige bærere af BRCA2-mutationer er livstidsrisikoen for cancer mammae beregnet til 6%, hvilket er 100 gange større end hos mænd i baggrundsbefolkningen. Mutationspositive mænd tilbydes på nuværende tidspunkt ingen overvågningsprogrammer. Bærere af BRCA1/2-mutationer har muligvis også en moderat øget risiko for andre cancersygdomme, herunder prostatacancer, pancreascancer og malignt melanom (15,16).
. Livstidsrisikoen for. cancer mammae har hos bærere af BRCA1-mutationer varierer mellem 56% og 87% i forskellige. undersøgelser (8–10). Den mediane alder på diagnosetidspunktet for cancer mammae er 42 år hos. BRCA1-mutationsbærere, hvilket er 20 år tidligere end hos patienter med sporadiske tumorer (11). Livstidsrisikoen for cancer ovarii varierer mellem 20 og 60% (10,12). BRCA2-mutationer er formentlig ansvarlige for en mindre del af arvelig disposition til cancer mammae. og cancer ovarii end BRCA1-mutationer. Den mediane alder på diagnosetidspunktet for cancer. mammae er 49 år, hvilket er højere end hos bærere af BRCA1-mutationer men lavere end for kvinder. med sporadisk cancer mammae (13). Livstidsrisikoen for cancer mammae varierer i forskellige. undersøgelser mellem 28 og 85% (14). Livstidsrisikoen for cancer ovarii er øget hos bærere af BRCA2- mutationer (10 – 27%). Hos mandlige bærere af BRCA2-mutationer er livstidsrisikoen for cancer. mammae beregnet til 6%, hvilket er 100 gange større end hos mænd i baggrundsbefolkningen. Mutationspositive mænd tilbydes på nuværende tidspunkt ingen overvågningsprogrammer. Bærere af BRCA1/2-mutationer har muligvis også en moderat øget risiko for andre cancersygdomme, herunder prostatacancer, pancreascancer og malignt melanom (15,16).")
15
PATIENT MED HØJPENETRANT MUTATION I BRCA1+2
70-80% Livstidsrisiko for brystkræft (BRCA1) 28-85% Livstidsrisiko for brystkræft (BRCA2) 6 % Livstidsrisiko for brystkræft hos mænd (BRCA2) Median alder ved diagnose 42 år (BRCA1) Median alder ved diagnose 49 år (BRCA2) 20-25% for brystkræft 2. gang i løbet af de følgende 10 år 10-60% Livstidsrisiko for c. ovarii. (BRCA 1) 10-27 %Livstidsrisiko for c. ovarii (BRCA2) Median alder nok som andre Øget risiko for prostatakræft (BRCA2) Brystkræft
 28-85% Livstidsrisiko for brystkræft (BRCA2) 6 % Livstidsrisiko for brystkræft hos mænd (BRCA2) Median alder ved diagnose 42 år (BRCA1) Median alder ved diagnose 49 år (BRCA2) 20-25% for brystkræft 2. gang i løbet af de følgende 10 år % Livstidsrisiko for c. ovarii. (BRCA 1) %Livstidsrisiko for c. ovarii (BRCA2) Median alder nok som andre. Øget risiko for prostatakræft (BRCA2) Brystkræft.")
16
OVERVÅGNING/REDUCERING VED KENDT HØJPENETRANT MUTATION/HBOC
Intensiveret overvågning af Mamma(e) Årlig MR, mammografi, UL Ovarier Årlig gyn. us med UL+ CA 125 Ingen kontrol af mænd Risikoreducerende kirurgi Mastectomi og rekonstruktion Salpingo-oophorectomi (c.ovarii samt c.mammae) Brystkræft
 Årlig MR, mammografi, UL. Ovarier. Årlig gyn. us med UL+ CA 125. Ingen kontrol af mænd. Risikoreducerende kirurgi. Mastectomi og rekonstruktion. Salpingo-oophorectomi (c.ovarii samt c.mammae) Brystkræft.")
17
PATIENT UDEN MUTATION Højrisiko familie men BRCA neg (>30%) for
C. mammae tilbydes årlig mammografi 30 – 50 år Screeningsmammografi 50 – 70 år C. mammae og c. ovarii tilbydes mammografi som ovenstående + kontrol af ovarier eller ooforektomi Moderat risiko familie (20-30%) for C. mammae tilbudt årlig mammografi 40 – 50 år Ikke forøget risiko familie(<20%) for C. mammae tilbydes screeningsmammografi år Bryst selv-undersøgelse anbefales ikke som screeningsmetode. • Mammografi anbefales som screeningsmetode til kvinder med forøget livstidsrisiko for cancer mammae (> 20%). Anbefalingerne vedrørende tidspunktet for første mammografi, intervallerne mellem screeningerne og mammografiens tekniske udførelse fremgår af Tabel 19.1. • MR-scanning kan anvendes som en komponent i den klinisk billeddiagnostiske mammaundersøgelse, men anbefales ikke som eneste screeningsmetode udenfor forsøgsprotokoller. • Profylaktisk mastektomi anbefales ikke, men ønsket om profylaktisk mastektomi efterkommes hos kvinder med en høj livstidsrisiko (>30%), der fastholder ønsket efter at have gennemført et genetisk rådgivningsforløb. • Salpingo-oophorectomi kan reducere risikoen for cancer mammae hos præmenopausale kvinder. Hos præmenopausale kvinder med lav eller moderat øget risiko (< 10%) anbefales salpingooophorectomi ikke. Kvinder med en høj livstidsrisiko (> 10%) bør oplyses om fordele og ulemper ved salpingo-oophorectomi. • Hormonal kontraception fører på kort sigt til en lille øgning af risikoen for cancer mammae hos kvinder i baggrundsbefolkningen. Samme effekt opnås formentlig hos kvinder med arvelig disposition til cancer mammae. Hormonal kontraception reducerer formentlig samtidig risikoen for cancer ovarii. Effekten på restlevetiden er ukendt. • Hormonal substitution efter menopausen frarådes generelt hos kvinder med høj (> 30%) eller moderat øget risiko for cancer mammae. • Det er uafklaret om Tamoxifen, Raloxifen eller andre østrogen receptor modulatorer reducerer risikoen for cancer mammae hos præmenopausale bærere af risikogivende BRCA1/2 mutationer. Tamoxifen reducerer risikoen for cancer mammae med 35 – 40% hos kvinder med moderat øget risiko (20 – 30%) for sygdommen. Det er dog uafklaret om reduktionen i risikoen for cancer mammae medfører en levetidsgevinst. Brystkræft
 for. C. mammae tilbudt årlig mammografi 40 – 50 år. Ikke forøget risiko familie(<20%) for. C. mammae tilbydes screeningsmammografi år. Bryst selv-undersøgelse anbefales ikke som screeningsmetode. • Mammografi anbefales som screeningsmetode til kvinder med forøget livstidsrisiko for cancer. mammae (> 20%). Anbefalingerne vedrørende tidspunktet for første mammografi, intervallerne. mellem screeningerne og mammografiens tekniske udførelse fremgår af Tabel • MR-scanning kan anvendes som en komponent i den klinisk billeddiagnostiske. mammaundersøgelse, men anbefales ikke som eneste screeningsmetode udenfor. forsøgsprotokoller. • Profylaktisk mastektomi anbefales ikke, men ønsket om profylaktisk mastektomi efterkommes hos. kvinder med en høj livstidsrisiko (>30%), der fastholder ønsket efter at have gennemført et genetisk. rådgivningsforløb. • Salpingo-oophorectomi kan reducere risikoen for cancer mammae hos præmenopausale kvinder. Hos præmenopausale kvinder med lav eller moderat øget risiko (< 10%) anbefales salpingooophorectomi. ikke. Kvinder med en høj livstidsrisiko (> 10%) bør oplyses om fordele og ulemper. ved salpingo-oophorectomi. • Hormonal kontraception fører på kort sigt til en lille øgning af risikoen for cancer mammae hos. kvinder i baggrundsbefolkningen. Samme effekt opnås formentlig hos kvinder med arvelig. disposition til cancer mammae. Hormonal kontraception reducerer formentlig samtidig risikoen for. cancer ovarii. Effekten på restlevetiden er ukendt. • Hormonal substitution efter menopausen frarådes generelt hos kvinder med høj (> 30%) eller. moderat øget risiko for cancer mammae. • Det er uafklaret om Tamoxifen, Raloxifen eller andre østrogen receptor modulatorer reducerer. risikoen for cancer mammae hos præmenopausale bærere af risikogivende BRCA1/2 mutationer. Tamoxifen reducerer risikoen for cancer mammae med 35 – 40% hos kvinder med moderat øget. risiko (20 – 30%) for sygdommen. Det er dog uafklaret om reduktionen i risikoen for cancer. mammae medfører en levetidsgevinst. Brystkræft.")
18
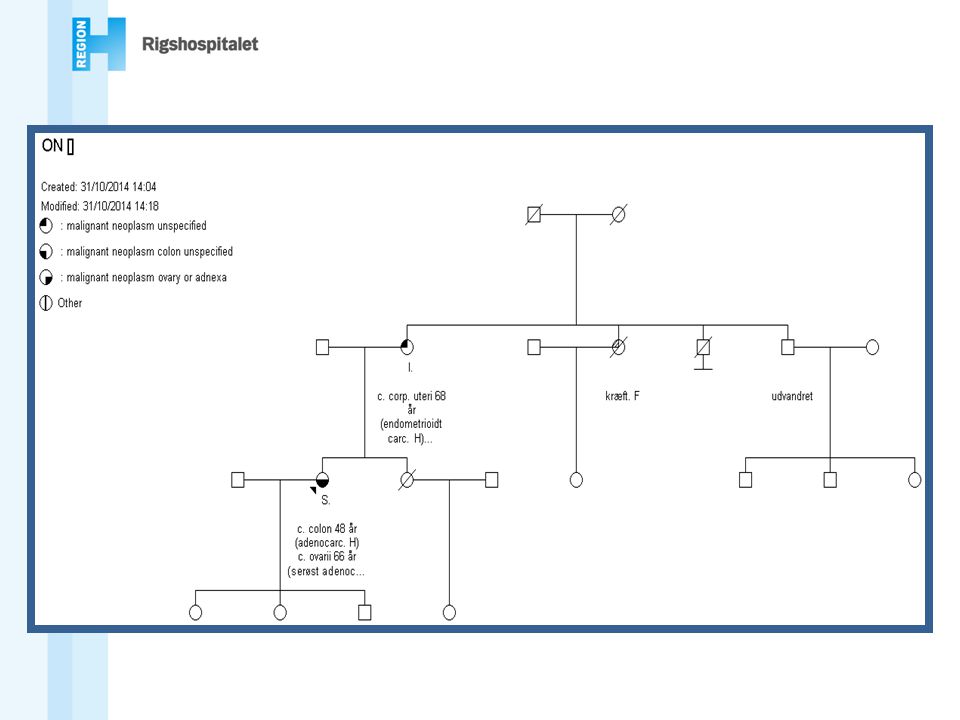
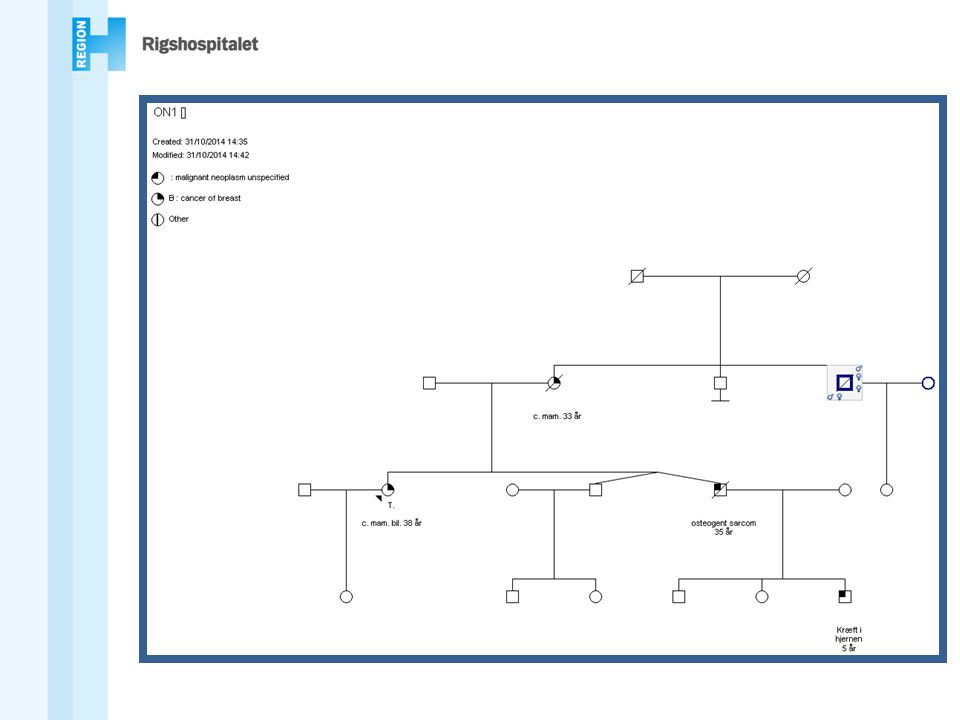
En person med brystkræft før 40 års alderen
Rekommandationer (DBCG) At genetisk udredning og rådgivning tilbydes i familier hvori der mistænkes en arvelig disposition til cancer mammae og/eller cancer ovarii, f.eks. familier hvor: En person med brystkræft før 40 års alderen En person med både bryst- og æggestokkræft To førstegradsslægtninge med brystkræft før 50 år eller æggestokkræft Tre førstegradsslægtninge med brystkræft over mindst to generationer, hvoraf mindst én fik påvist sygdommen før 50 års alderen En mand med brystkræft En person i hvis familie der forekommer en mutation, der medfører øget risiko for bryst- og/eller æggestokkræft Bilateral brystkræft Triple negative cancer under 50 år Flere tumorer i samme bryst under 50 år Mette Wilhjelm
 At genetisk udredning og rådgivning tilbydes i familier hvori der mistænkes en arvelig disposition til cancer mammae og/eller cancer ovarii, f.eks. familier hvor: En person med brystkræft før 40 års alderen. En person med både bryst- og æggestokkræft. To førstegradsslægtninge med brystkræft før 50 år eller æggestokkræft. Tre førstegradsslægtninge med brystkræft over mindst to generationer, hvoraf mindst én fik påvist sygdommen før 50 års alderen. En mand med brystkræft. En person i hvis familie der forekommer en mutation, der medfører øget risiko for bryst- og/eller æggestokkræft. Bilateral brystkræft. Triple negative cancer under 50 år. Flere tumorer i samme bryst under 50 år. Mette Wilhjelm.")
19
GENETISK RÅDGIVNING SOM HASTESAG FOR AT MÅLRETTE BEHANDLINGEN
Lumpectomeret uden lymfeknude metastaser (eventuelt undgå strålebehandling hvis profylaktisk mastectomi) Neoadjuverende behandling (eventuelt få fjernet andet bryst hvis profylaktisk mastectomi) Rekonstruktion (eventuelt få lavet rekonstruktion af det andet bryst ) Brystkræft
 Neoadjuverende behandling. (eventuelt få fjernet andet bryst hvis profylaktisk mastectomi) Rekonstruktion. (eventuelt få lavet rekonstruktion af det andet bryst ) Brystkræft.")
20
FOUNDER MUTATION Relativ høj hyppighed af en bestemt genotype.
En eller få bærere af genotypen er ophav til en stor del af mennesker i en population. Eksempelvis grønlændere og Ashkenazi jøder. Grønlænder blive testet på grønland Hvis en grønlænder DK får bc så kan de henvises til genetisktest specielt få lavet foundermutationstest

21
Mette Wilhjelm

22
Mette Wilhjelm

23
Mette Wilhjelm

24
Mette Wilhjelm

25
HNPCC: Hereditær Nonpolypøs Colorektal Cancer
Lynch syndrom / HNPCC: Hereditær Nonpolypøs Colorektal Cancer Susanne Hein Klinisk Genetisk Klinik Rigshospitalet

26
KRITERIER FOR HENVISNING
En person med tarmkræft < 50 år En person med både tarmkræft og livmoderkræft To førstegradsslægtninge med Lynch syndrom relateret kræft (tyk / ende / tyndtarm, livmoder, æggestok, nyrebækken, urinleder) hvoraf mindst en er diagnostiseret før < 50 år FAP fam. /anden arvelig sgd . tarmkræft
 hvoraf mindst en er diagnostiseret før < 50 år. FAP fam. /anden arvelig sgd . tarmkræft.")
27
Genetisk risikovurdering
og rådgivning Genetisk testing Surveillance Risikoreducerende kirurgi

28
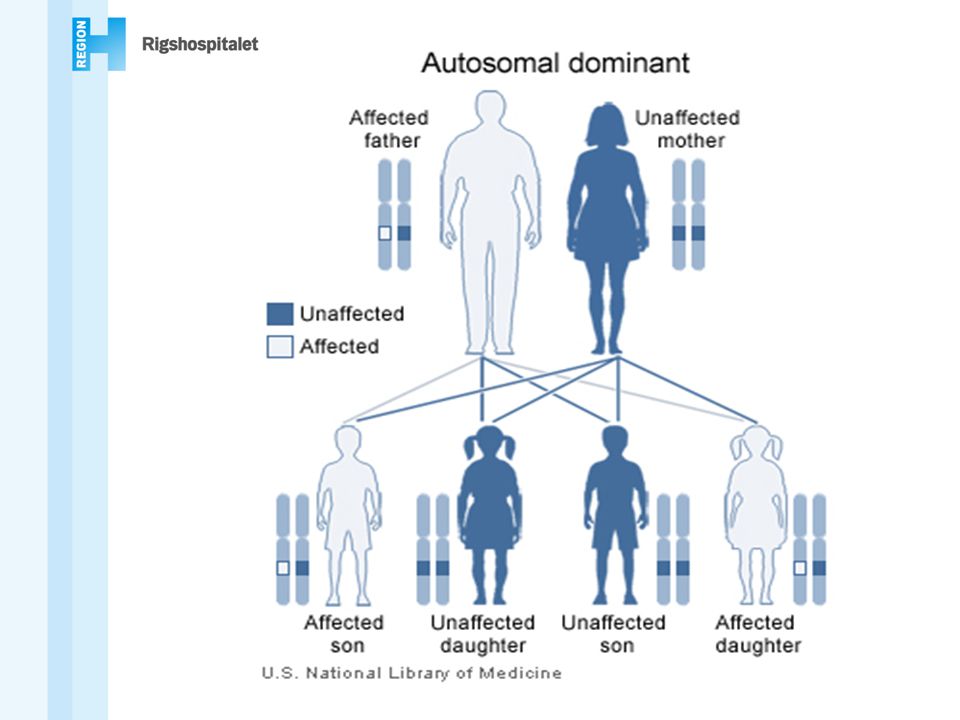
ARVELIG CANCER Autosomal dominant Autosomal recessiv
Hereditary breast-ovarian cancer HBOC Cowden syndrom Familiær colonpolypose (FAP) Gorlin syndrom Hereditary non polyposis colorectal cancer (HNPCC, Lynch syndrome) Li Fraumeni syndrom Diffus ventrikelcancer (HDGC) Malignt melanom syndrom Multipel endokrin neoplasi 1,2 Neurofibromatose 1,2 Prostatacancer syndromer Retinoblastom von Hippel Lindau sygdom Autosomal recessiv Ataxia telangiectasia Bloom syndrom Cockayne syndrom Fanconi anæmi Nijmegen breakage syndrom Severe combined immuno deficiency (SCID) Werner syndrom Xeroderma pigmentosum MYH polypose Kønsbunden recessive Wiscott Aldridge syndrom
 Gorlin syndrom. Hereditary non polyposis colorectal cancer (HNPCC, Lynch syndrome) Li Fraumeni syndrom. Diffus ventrikelcancer (HDGC) Malignt melanom syndrom. Multipel endokrin neoplasi 1,2. Neurofibromatose 1,2. Prostatacancer syndromer. Retinoblastom. von Hippel Lindau sygdom. Autosomal recessiv. Ataxia telangiectasia. Bloom syndrom. Cockayne syndrom. Fanconi anæmi. Nijmegen breakage syndrom. Severe combined immuno deficiency (SCID) Werner syndrom. Xeroderma pigmentosum. MYH polypose. Kønsbunden recessive. Wiscott Aldridge syndrom.")
30
Hereditary breast cancer
Arvelig cancer - germline mutation Sporadisk cancer - somatisk mutation Hereditary breast cancer Somatiske forældreceller Kønsceller Somatiske celler hos børn Tumorcelle Tumorcelle

31
COLORECTAL CANCER - GENETICS

32
GENETISK TESTNING Diagnostik Behandling Prædiktiv gentest: -mutation: populations cancerrisiko +mutation: høj cancerrisiko - follow up Hvis mutation ikke påvises: stamtræ basis for risikovurdering follow up af alle nære slægtninge

33
MMR gener: MLH1, MSH2, MSH6, PMS2 MSI / IHC

34
Afficerede slægtninge:
KLINISK OPFØLGNING Afficerede slægtninge: - kirurgi - kemoterapi Raske slægtninge: - screening/surveillance - risikoreducerende kirurgi - medicinsk behandling Prænatal/præimplantationsdiagnostik (PGD)
")
35
KLINISK OPFØLGNING – LYNCH SYNDROM
Koloskopi hvert 2. år fra 25 år Gynækologisk US hvert 2. år fra 35 år incl. vaginal UL og biopsi KUN Lynch syndrom familier +mutation Nyre UL i familier med urothelcelletumorer

36
Cancer livstidsrisiko – Lynch syndrom
Colorektal cancer* % (♂>♀) Metakron CRC % efter år Endometriecancer 60% (MSH6>MSH2>MLH1) Ovariecancer ca. 10% *højresidig, mucinøs
 Metakron CRC 40-50% efter år. Endometriecancer 60% (MSH6>MSH2>MLH1) Ovariecancer ca. 10% *højresidig, mucinøs.")
37
Cumulative Risks of Cancer by Age
Lynch syndrome Bonadona, V. et al. JAMA 2011;305:

38
Bonadona, V. et al. JAMA 2011;305:2304-2310

39
Anden klinisk opfølgning
Klassisk HNPCC. Amsterdam I kriterier: 3 fam. medl. Med kræft i tyktarm i 2. generationer. En af de tre er 1. gradsslægtninge til de to andre. En af de tre er < 50 år. Amsterdam II kriterier: som I, men med kræft i livmoder, tyndtarm el. øvre urinveje

40
HNCC sandsynlig: To personer med tarmkræft i lille fam. En < 50 år
To personer med tarmkræft, og en med adenom (svær dysplasi) el. HNPCC rel. kræft. Tre personer med tarmkræft i lille fam. En < 50 år, og de er ikke 1. grads slægtninge
 el. HNPCC rel. kræft. Tre personer med tarmkræft i lille fam. En < 50 år, og de er ikke 1. grads slægtninge.")
41
HNPCC late onset: Som Amsterdam I kriterier, men alle personer er > 50 år Moderat øget risiko: En i fam. har fået tarmkræft før 50 år To personer, der er 1. grads slægtninge, har fået tarmkræft efter 50 år.

42
Klassisk HNPCC, koloskopi hvert 2 år fra 25 års alderen
Klassisk HNPCC, koloskopi hvert 2 år fra 25 års alderen. Late onset HNPCC, koloskopi hvert 2 år fra 45 års alderen. Moderat risiko, koloskopi hvert 5 år. Fra 10 år før diagnosealder for yngste tilfælde (dog senest fra 50 år)
")
44
Flossman and Rothwell Lancet 2007
People who took part in the early cardiovascular aspirin trials had fewer colorectal cancers 10 years later 10% 8% No treatment 6% Log rank p=0.003 4% Aspirin 2% 0% 5 10 15 20 25 30 Length of follow-up (years) Flossman and Rothwell Lancet 2007
 Flossman and Rothwell Lancet")
45
Arvelig kræftform Øget risiko for kræft i disse organer Gener Bryst- og æggestokkræft Bryst, æggestokke og prostata BRCA1 BRCA2 RAD51C Familiær colonpolypose (FAP) Tarm APC Hereditær nonpolypøs colorektal cancer HNPCC (Lynch syndrom) Tarm, livmoder, urinveje og æggestokke MLH1 MSH2 MSH6 (PMS2) Kræft i mavesækken Mavesækken (diffus type) og bryst CDH1 Modermærkekræft Modermærke og bugspytkirtel CDKN2A CDK4 MYH-associeret tarmkræft MYH Retinoblastom (øjensvulst) Øjne og knogler RB1 Prostatakræft Prostata Mange gener, men endnu ikke anvendeligt til gentest Er der f.eks. arvelig bryst- og æggestokkræft i familien (kolonne1), kan man i den pågældende familie se en øget forekomst af både brystkræft, æggestokkræft og prostatakræft (kolonne 2). I kolonne 3 kan man se navnet på det/de gen(er), hvori mutationer går i arv fra den ene person til den anden.
 Tarm. APC. Hereditær nonpolypøs colorektal cancer HNPCC (Lynch syndrom) Tarm, livmoder, urinveje og æggestokke. MLH1 MSH2 MSH6 (PMS2) Kræft i mavesækken. Mavesækken (diffus type) og bryst. CDH1. Modermærkekræft. Modermærke og bugspytkirtel. CDKN2A CDK4. MYH-associeret tarmkræft. MYH. Retinoblastom (øjensvulst) Øjne og knogler. RB1. Prostatakræft. Prostata. Mange gener, men endnu ikke anvendeligt til gentest. Er der f.eks. arvelig bryst- og æggestokkræft i familien (kolonne1), kan man i den pågældende familie se en øget forekomst af både brystkræft, æggestokkræft og prostatakræft (kolonne 2). I kolonne 3 kan man se navnet på det/de gen(er), hvori mutationer går i arv fra den ene person til den anden.")
46
Arvelige sygdomme Øger risikoen for kræft i disse organer Gener Peutz Jeghers syndrom Tarm og bryst STK11 Juvenil polypose Tarm SMAD4 BMPR1A Li-Fraumenis syndrom Bryst, bindevæv/knogler, binyre og hjerne TP53 Gorlins syndrom Almindelig hudkræft PTCH Von Hippel Lindaus sygdom Karsvulster i hjerne og rygmarven (ikke ondartet), nyre og binyre VHL Multiple endokrin neoplasi MEN2 Skjoldbruskkirtel, binyre RET Birth-Hogg Dube syndrom Nyre FLCN Neurofibromatose 1 Bindevæv, binyre NF1 Tuberøs sklerose Bindevæv, nyre TSC1 TSC2 Ataxia telangiectasi Blod ATM Blooms syndrom Hud, blod og nyre BLM Fanconis anæmi Blod og hud Mange gener, bl.a. BRCA2 Xeroderma pigmentosum Hud og blod XP-A,-B,-C,-D,-E,-F,-G Nijmegen breakage syndrom Blod, rygmarv og bindevæv NBS1 Er der f.eks. Peutz Jeghers syndrom i familien (kolonne 1), kan man i den pågældende familie se en øget forekomst af både tarm- og brystkræft (kolonne 2). I kolonne 3 kan man se navnet på det/de gen(er), hvori mutationer går i arv fra den ene person til den anden.
, nyre og binyre. VHL. Multiple endokrin neoplasi MEN2. Skjoldbruskkirtel, binyre. RET. Birth-Hogg Dube syndrom. Nyre. FLCN. Neurofibromatose 1. Bindevæv, binyre. NF1. Tuberøs sklerose. Bindevæv, nyre. TSC1 TSC2. Ataxia telangiectasi. Blod. ATM. Blooms syndrom. Hud, blod og nyre. BLM. Fanconis anæmi. Blod og hud. Mange gener, bl.a. BRCA2. Xeroderma pigmentosum. Hud og blod. XP-A,-B,-C,-D,-E,-F,-G. Nijmegen breakage syndrom. Blod, rygmarv og bindevæv. NBS1. Er der f.eks. Peutz Jeghers syndrom i familien (kolonne 1), kan man i den pågældende familie se en øget forekomst af både tarm- og brystkræft (kolonne 2). I kolonne 3 kan man se navnet på det/de gen(er), hvori mutationer går i arv fra den ene person til den anden.")
49
Genes, genes everywhere…

Lignende præsentationer